The Spectrochemical
Series
One of the important aspects of CFT is that all ligands are not
identical when it comes to causing a separation of the energy of the
d-orbitals. For transition metal compounds, there is clear
evidence for this from the multitude of colours available for a
given metal ion when the ligands or stereochemistry are varied.
In octahedral complexes, this can be considered a reflection of
the energy difference between the higher dz2,
dx2-y2(eg subset) and the dxy,
dyz, dxz (t2g subset).
It has been established that the ability of ligands to cause a
large splitting of the energy between the orbitals is essentially
independent of the metal ion and the SPECTROCHEMICAL SERIES is a
list of ligands ranked in order of their ability to cause large
orbital separations. Thus halides cause a small splitting and CO
gives a large splitting.
A shortened list includes:
I- < Br- < SCN- ~Cl- < F- <
OH- ~ ONO- < C2O42-
< H2O
< NCS- < EDTA4- < NH3 ~
pyr ~ en < bipy < phen < CN- ~ CO
From a purely ionic basis we would expect CO < H2O
< C2O42- <
EDTA4-
i.e. that the most negatively charged species should interact the
most strongly.
That this is not the case is a reflection of covalent
interactions and is a limitation of the CFT ionic model.
Ligand Field Theory
Ligand Field Theory can be considered an extension of Crystal
Field Theory such that all levels of covalent interactions can be
incorporated into the model.
Treatment of the bonding in LFT is generally done using
Molecular Orbital Theory. A qualitative approach that can be used
for octahedral metal complexes is given in the following 3
diagrams.
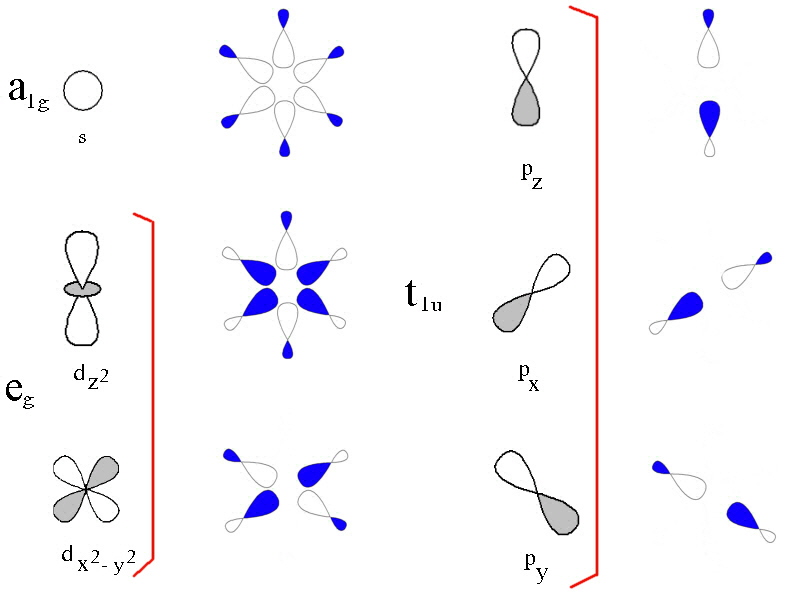
In the first diagram, the 3d, 4s and 4p metal ion atomic orbitals are shown together
with the ligand group orbitals that would have the correct symmetry to be
able to overlap with them.
The ligand group orbitals are generated by taking 6 sigma orbitals from the
ligands, designated as σx, σ-x,
σy, σ-y, σz,
σ-z and then combining them to make 6 ligand group orbitals.
(labelled eg, a1g, t1u)
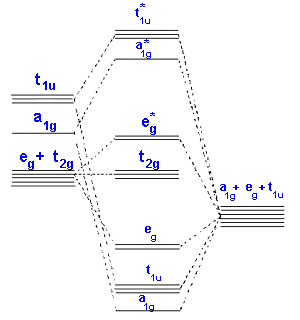
In the second diagram only sigma bonding is considered and
it shows the combination of the metal 3d, 4s and 4p orbitals
with OCCUPIED ligand group orbitals (using 1 orbital from each ligand).
The result is that that the metal electrons would be fed into t2g and eg*
molecular orbitals which is similar to the CFT model
except that the eg orbital is now eg*.
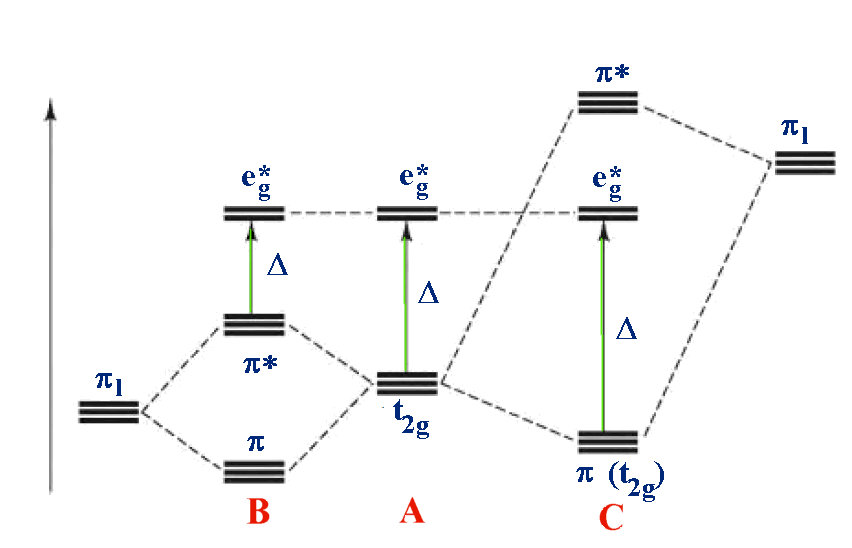
For example: B - [M(II)I
6]
4-
A - [M(II)(H
2O)
6]
2+
C - [M(II)(CN)
6]
4-
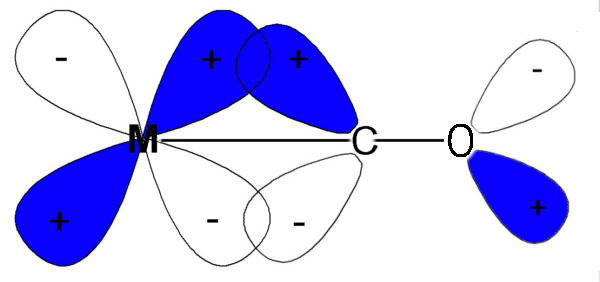
Case A is the same as above, ignoring π interactions.
For case B, the ligand π orbitals are full
and at lower energy than the metal t2g. This causes a decrease
in the size of Δ.
For case C, the ligand π orbitals are empty
and at higher energy than the metal t2g. This causes an increase
in the size of Δ.
Returning to the problem of correctly placing ligands in the
Spectrochemical series, the halides are examples of case A and
groups like CN- and CO are examples of case B. It is possible
then to explain the Spectrochemical series once covalent effects
are considered.
Some convincing arguments for covalency and effects on Δ come from
a study of the IR spectra recorded for simple carbonyl compounds
e.g. M(CO)6.
The CO molecule has a strong triple bond which in the IR gives rise to a
strong absorption at ~2140 cm-1.
For the series [Mn(CO)6]+, [Cr(CO)6] and
[V(CO)6]-, which are
isoelectronic, the IR bands for the CO have shifted to 2090, 2000
and 1860 cm-1 respectively. Despite the fact that the metals have the
same number of electrons (isoelectronic) the frequency of force constant of
the CO bond is seen to vary Mn+ > Cr > V-.
This can not be explained on an ionic basis but is consistent with
the π bonding scheme since the greater the positive charge on the metal,
the less readily the metal can delocalise electrons back into the π*
orbitals of the CO group.
Note that the IR values we are dealing with relate to the CO bond and
not the M-C so when the CO frequency gets less then it is losing triple bond
character and becoming more like a double bond. This is expected if electrons are
pushed back from the metal into what were empty π* antibonding orbitals.
return to the CHEM2101 (C21J) course
outline
 Return to Chemistry, UWI-Mona,
Home Page
Return to Chemistry, UWI-Mona,
Home Page
Copyright © 2000-2010 by Robert John
Lancashire, all rights reserved.
Created and maintained by Prof. Robert J.
Lancashire
The Department of Chemistry, University of the West
Indies,
Mona Campus, Kingston 7, Jamaica.
Created September 2000. Links checked and/or
last modified 13th September 2010.
URL
http://wwwchem.uwimona.edu.jm/courses/LFT.html