Preparation of some Werner Complexes
Experimental
Cobalt sulfate
cis-tetraamminecarbonatocobalt(III) ion
7g of ammonium carbonate is dissolved in 20 mL of water and 20 mL
of concentrated ammonia solution is added. While stirring, this
is then added to a solution containing 5g cobalt sulfate in 10 mL
of water. 3 mL of 30% hydrogen peroxide is then slowly added.
(Handle H2O2 carefully. If spilled on the skin, wash the
affected area immediately with water; hydrogen peroxide can cause
severe skin burns).Next, pour the solution into an
evaporating dish in a fumehood and concentrate it over a gas
burner to 30-35 mL. Do not allow the solution to boil. During the
evaporation add, in small portions, 2g of ammonium carbonate.
Suction filter (with water aspirator) the hot solution and then
cool the filtrate in an ice bath. Under suction, filter off the
red product crystals of [CoCO3(NH3)4]2SO4.3H2O. Wash the product
in the filtration apparatus using a saturated solution prepared
from a small portion of the precipitate.
Record the yield and show the product to your demonstrator.
cis-tetraamminediaquacobalt(III) ion
2g of the previously prepared tetraamminecarbonatocobalt(III)
complex is dissolved in 55mL of 0.3M sulfuric acid. Evolution of
carbon dioxide should occur. The clear solution is then treated
with 20-25 mL of ethanol, added in small portions. The
precipitate is filtered off, washed with 50% ethanol/water until
free of acid. (Check with litmus paper.) Finally, the product is
dried in air. Record the yield and show the product to your
demonstrator.
"hexol"
2g of the previously prepared tetraamminediaquacobalt(III)
complex is dissolved in 25-30mL of 0.2M ammonia solution. After
24 hour (can be left until the next session) purple-black
crystals precipitate. These are suction filtered. Record the
yield and show the product to your demonstrator.
Cobalt chloride
hexaamminecobalt chloride
3g of cobalt(II) chloride hexahydrate is dissolved in a boiling
solution of 2g of ammonium chloride in 5mL of water. This is then
added to 0.15g of decolourising charcoal in a 100 mL flask and
cooled by running water on the outside of the flask. The original
vessel is rinsed with 8 mL of concentrated ammonia and these
washings are added to the flask with the charcoal. The flask is
then cooled to 10°C or below.
8 mL of "20 volume" hydrogen peroxide is then slowly added in
portions and the falsk is briskly shaken. When all the oxidising
agent has been added, the mixture is heated gradually to 60°C and
maintained at this temperature until the pinkish tint in the
liquid disappears (about 30 minutes). During this time the flask
should be shaken frequently. At the end of the heating period,
crystals should start to separate.
The flask is then cooled in an ice-bath for about 10 minutes and
the crude solid filtered off via a Buchner funnel. Portions of
the filtrate are used to rinse and solid remaining in the flask
on to the filter. The product is allowed to drain for 5 minutes
and then is recrystallised as follows:
Transfer the product to a beaker containing a boiling mixture of
25 mL of water and 1 mL of concentrated hydrochloric acid. When
all the solid (except the charcoal) has dissolved, filter the hot
liquid, via a Buchner funnel. 3 mL of concentrated hydrochloric
acid is then added to the filtrate and the solution cooled in an
ice-bath.
The golden brown crystal of the hexaamminecobalt(III) ion should
be obtained in high yield. Filter off the crystals on a sintered
glass filter funnel and wash them with ethanol (NO FLAMES).
Transfer the crystals to a small weighed beaker and dry them in
an oven at 110-120 for 30 minutes. (NOTE that higher temperatures
will decompose the product). Allow the crystals to cool in a
dessicator and then record the yield and show the product to your
demonstrator.
pentaamminechlorocobalt(III) chloride
In a fume hood, dissolve 1 g of ammonium chloride in 9 ml
concentrated aqueous ammonia in a 100-ml Erlenmeyer flask. (The
combination of NH4Cl and NH3 (aq.)
guarantees a large excess of the NH3 ligand.) Stir the
ammonium chloride solution vigorously while adding 2 g of finely
divided CoCl2.6H2O in small portions. A
yellow-pink precipitate of the hexaammine Co(II) salt forms on
slight warming as the reddish starting material dissolves. Any
air oxidation that occurs during this exothermic stage is ignored
since the solution is going to be fully oxidised by adding
hydrogen peroxide.
Caution: 30% hydrogen peroxide is a strong oxidizing agent that
will cause severe burns and bleaching of skin and clothing.
Slowly add 2 mL 30% hydrogen peroxide to the brown Co slurry,
using a burette. An addition rate of about 2 drops per second is
usually sufficient, but care should be taken to avoid excessive
effervescence. (If the reaction shows signs of excessive
effervescence, stopping the stirring momentarily will usually
prevent overflow of the solution.)
You should notice that all the Co(II) ammine dissolves to form a
deep red solution. (This corresponds to the formation of the
pentaammineaquacobalt(III) salt.)
When the effervescence has virtually ceased, cautiously add 6 mL
of concentrated HCl in small portions and with continuous
stirring. This operation needs to be carried out in a fume hood
since fumes of ammonium chloride will be produced during the
neutralisation. After this point the reaction may be removed from
the hood. A purple product should then precipitate from the hot
reaction mixture leaving a pale green-blue supernatant liquid.
While occasionally stirring, use a steam bath or hot plate to
heat the solution to 60°C. Hold the temperature between
55°C and 65°C for 15 min; this incubation period is
necessary to allow complete displacement of all aqua ligands.
Collect the purple product by filtration through a No. 3 sinter
glass crucible. The mother liquor may be discarded.
When the product has been drained well it is washed with 4 mL
ice-cold deionised water in small portions, followed by 5 mL
ice-cold 95% ethanol. (The solutions must be cold to prevent
undue loss of product by redissolving.) Transfer the product to a
crucible and dry in an oven at 100°C for one hour. This helps
complete the conversion of any remaining
pentaammineaquacobalt(III) salt. Submit your sample to the demonstrator.
pentaamminenitritocobalt(III) ion
Dissolve 1g of pentaamminechlorocobalt(III) chloride, by gently
warming and stirring, in a mixture of 20 mL water and 1.5 mL of
concentrated ammonia solution (CARE! EYES!). Filter off any
slight precipitate of cobalt oxide that may form, and then cool
the filtrate in ice to about 10°C. When cool, add 2M HCl dropwise
until the solution is just neutral to litmus.
Next, dissolve 1g of sodium nitrite in the solution before
adding 0.5 mL 11 M HCl. Allow the solution to stand in an
ice-bath for 1 hour and then filter off the salmon-pink crystals.
Wash with 5 mL of ice-water, followed by 5 mL of alcohol, and air
dry on filter paper at room temperature. Record the yield and
show the product to your demonstrator.
pentaamminenitrocobalt(III) ion
Dissolve 0.5g of the above nitrito-complex in 4 mL of hot water
containing a few drops of concentrated ammonia solution and then
add, while cooling, 4 mL of concentrated HCl. Cool the solution
in an ice-bath and filter off the yellow-brown crystals. Wash the
product with 2 mL of alcohol and air dry on filter paper at room
temperature. Record the yield and show the product to your demonstrator.
linkage isomerism
These latter two complexes are examples of what is described as
linkage isomerism. The initial reaction requires the formation of
the aqua complex as intermediate and then the replacement of the
OH2 by the O-bonded nitrito ligand.
This replacement is unexpectedly labile considering the
usual inert reactions of Co(III). A detailed isotopic study
revealled that the Co-O bond remained intact with the bonds
on the O being substituted rather than at the Co.
The second reaction where the O-bonded group is replaced
by the N-bonded nitro group does proceed relatively slowly
as expected for a Co-O to Co-N substitution.
The IR of these linkage isomers
is available. The O-bonded isomer will slowly convert to the
more stable N-bonded isomer with time. It is possible to study
this reaction via IR by preparing a KBr pellet and recording
the IR spectrum over a period of a few days.
 Return to Chemistry, UWI-Mona,
Home Page
Return to Chemistry, UWI-Mona,
Home Page
Copyright © 2006 by Robert John
Lancashire, all rights reserved.
Created and maintained by Prof. Robert J.
Lancashire,
The Department of Chemistry, University of the West Indies,
Mona Campus, Kingston 7, Jamaica.
Created Sept 1995. Links checked and/or last
modified 10th September 2006.
URL
http://wwwchem.uwimona.edu.jm/lab_manuals/Werner2.html
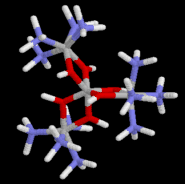 hexol.
hexol.