Lecture 5c. Structure of the elements, continued P, S and I.
Elemental phosphorus
exists in a number of different allotropes.
White phosphorus
The most important form of elemental phosphorus from the perspective of
applications and the chemical literature is white phosphorus.
It consists of tetrahedral P4 molecules,
in which each atom is bound to the other three atoms by a single bond.
This P4 tetrahedron is also present in liquid and gaseous phosphorus
up to the temperature of 800 °C when it starts decomposing to P2 molecules.
Solid white phosphorus exists in two forms. At low-temperatures, the β form is stable.
At high-temperatures the α form is predominant. These forms differ in terms
of the relative orientations of the constituent P4 tetrahedra.
The history of the match is linked to the discovery of the
allotropes of phosphorus.
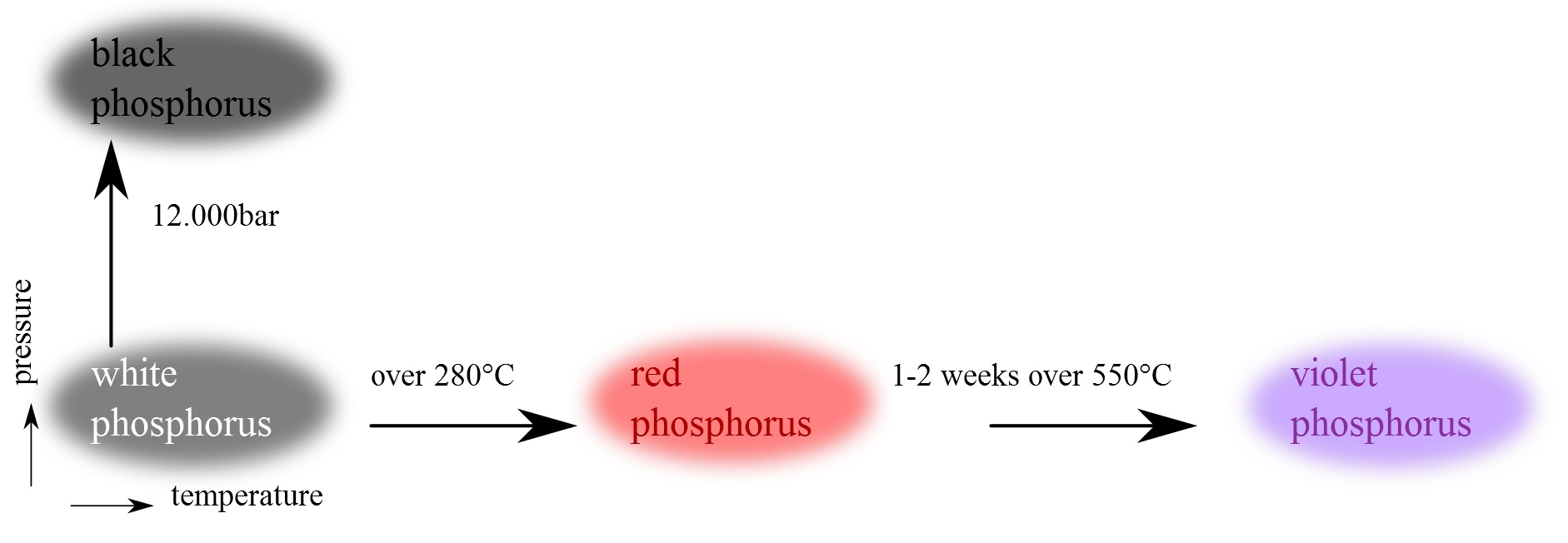
Allotropes of Phosphorus
white
|
red
|
violet - Hittorf
|
black
|
White phosphorus is the most reactive, the least stable, the most volatile,
the least dense, and the most toxic of the allotropes. White phosphorus
gradually changes to red phosphorus. This transformation is accelerated
by light and heat, and samples of white phosphorus almost always contain
some red phosphorus and accordingly appear yellow. For this reason,
white phosphorus that is aged or otherwise impure is sometimes called
yellow phosphorus. White phosphorus glows
in the dark (when exposed to oxygen) with a very faint tinge of green and blue,
is highly flammable and pyrophoric (self-igniting) upon contact with air and
is toxic (causing severe liver damage on ingestion). Owing to its pyrophoricity,
white phosphorus has been used as an additive in napalm. The odour of combustion of
this form has a characteristic garlic smell, and samples are commonly coated
with white "phosphorus pentoxide", which consists of P4O10
tetrahedra with oxygen inserted between the phosphorus atoms and at
their vertices. White phosphorus is insoluble in water but soluble in
carbon disulfide.
Red phosphorus
In 1847
Anton von Schrotter found that sunlight changed white/yellow into
red phosphorus, even when moisture and atmospheric oxygen were rigorously excluded.
The red product was separated from the residual yellow phosphorus by treatment
with carbon disulfide. Red phosphorus was also prepared from the yellow variety by
heating it to about 250 °C. in an inert gas. Heating to higher temperatures
reconverted the red modification to the yellow one.
Red phosphorus exists as an amorphous network and does not ignite in air
at temperatures below 240 °C.
Violet phosphorus
In 1865, Johann Hittorf heated
red phosphorus in a sealed tube at 530 °C. The upper part of the tube was
kept at 444 °C. Brilliant opaque monoclinic, or rhombohedral, crystals sublimed.
This form is sometimes known as "Hittorf's phosphorus" (or violet
or α-metallic phosphorus).
Black phosphorus
Black phosphorus is the thermodynamically stable form of phosphorus at
room temperature and pressure. It is obtained by heating white phosphorus under
high pressures (12,000 atmospheres). In appearance, properties and structure
it is similar to graphite,
being black and flaky, a conductor of electricity, and having puckered sheets
of linked atoms.
Black phosphorus has an orthorhombic structure and is the least reactive allotrope:
a result of its lattice of interlinked six-membered rings.
Each atom is bonded to three other atoms.
No other element forms more solid allotropes than sulfur. At present,
about 30 well characterized sulfur allotropes are known of which the most common
form found in nature is the greenish-yellow orthorhombic α-sulfur,
containing puckered rings of S8.
α-sulfur
When pure it has a greenish-yellow colour (traces of cyclo-S7 in
commercially available samples make it appear yellower). It is practically
insoluble in water and is a good electrical insulator with poor thermal
conductivity. It is quite soluble in carbon disulfide: 35.5 g/100 g solvent
at 25 °C. It has a rhombohedral crystal structure. This is the predominant
form found in "flowers of sulfur", "roll sulfur" and "milk of sulfur".
It contains S8 puckered rings, alternatively called a crown shape. The S-S
bond lengths are all 206 pm and the S-S-S angles are 108° with a
dihedral angle of 98°. At 95.3 °C, α-sulfur converts
to β-sulfur.
β-sulfur
This is a yellow solid with a monoclinic crystal form and is less dense
than α-sulfur. Like the α- form it contains puckered S8 rings and
only differs from it in the way the rings are packed in the crystal.
It is unusual because it is only stable above 95.3 °C, below this it
converts to α-sulfur. It can be prepared by crystallising
at 100 °C and cooling rapidly to slow down formation of α-sulfur.
It has a melting point of about 120 °C and decomposes at around this
temperature.
γ-sulfur
This form, first prepared by F.W Muthmann in 1890, is sometimes
called "nacreous sulfur" or "mother of pearl sulfur" because of its
appearance. It crystallises in pale yellow monoclinic needles.
It contains puckered S8 rings like α-sulfur and β-sulfur and only
differs from them in the way that these rings are packed. It is
the densest form of the three. It can be prepared by slowly cooling
molten sulfur that has been heated above 150 °C or by chilling
solutions of sulfur in carbon disulfide, ethyl alcohol or hydrocarbons.
It is found in nature as the mineral
rosickyite.
Some allotropes of Sulfur
S6 - cyclohexasulfur
|
α-S8
|
S12 - cyclododecasulfur
|
S6 - cyclo-hexasulfur
This was first prepared by M.R. Engel in 1891 who reacted HCl with
thiosulfate, HS2O3-. Cyclo-S6 is
orange-red and forms rhombohedral crystals. It is called ρ-sulfur,
ε-sulfur, Engel's sulfur and Aten's sulfur. Another method of
preparation involves reacting a polysulfane with sulfur monochloride:
H2S4 + S2Cl2 →
cyclo-S6 + 2 HCl (dilute solution in diethyl ether)
The sulfur ring in cyclo-S6 has a "chair" conformation, reminiscent
of the chair form of cyclohexane. All of the sulfur atoms are equivalent.
Cyclo-dodecasulfur
Thermodynamically, S12 is the second most stable sulfur ring after S8. Therefore,
S12 is formed in many chemical reactions in which elemental sulfur is a
product. In addition, S12 is a component of liquid sulfur at all temperatures.
The same holds for S18 and S20 which are often formed together with S12.
Its structure can be visualised as having sulfur atoms in three parallel planes,
3 in the top, 6 in the middle and three in the bottom.
Liquid sulfur after equilibration contains sulfur homocycles of all sizes
and some of these can be isolated by quenching, extraction, fractional
precipitation and crystallization depending on their differing solubilities.
Cyclo-S12 can be prepared by heating elemental sulfur to about 200 °C for
5-10 min and then allowing the mixture to cool to 140-160 °C within about
15 min. Once the melt has become less viscous, it is poured in as thin
a stream as possible into liquid nitrogen in order to quench the equilibrium.
Recrystallization of the yellow powder from CS2 allows the isolation
of an adduct which slowly loses the solvent to give the cyclo-dodecasulfur.
Note that both B and S form stable E12 species but the structures
(and coordination numbers) are quite different.
Iodine was discovered by French chemist Bernard Courtois in 1811.
His father was a manufacturer of saltpeter (a vital part of gunpowder) and
at the time of the French Napoleonic Wars, saltpeter was in
great demand. Saltpeter produced from French niter beds required sodium
carbonate, which could be isolated from seaweed collected on the coasts of
Normandy and Brittany. To isolate the sodium carbonate, the seaweed was burned
and the ash washed with water. The remaining waste was destroyed by adding
sulfuric acid. Courtois once added excessive sulfuric acid and a cloud of
purple vapour rose. He noted that the vapour crystallized on cold surfaces,
making dark crystals. Courtois suspected that this was a new element but
lacked the funds to pursue it further.
Samples of the material reached Humphry Davy and Joseph Louis Gay-Lussac
and in early December 1813 both claimed that they had identified a new element.
Arguments erupted between them over who had identified iodine first, but
both scientists acknowledged Courtois as the first to isolate the element.
Iodine is found on Earth mainly as the highly water-soluble iodide ion
I-, concentrated in oceans and brine pools. Like the
other halogens, free iodine occurs mainly as a diatomic molecule I2.
In the universe and on Earth, iodine's high atomic number makes it a relatively
rare element. However, its presence in ocean water has given it a role in
biology. It is the heaviest essential element widely utilized by life in
biological functions.
Under standard conditions, iodine is a bluish-black solid that sublimes
to form a noxious violet-pink gas. It melts at 113.7 °C (386.85 K) and forms compounds
with many elements but is less reactive than the other halogens, and has some
metallic light reflectance.
Elemental iodine is slightly soluble in water, with one gram dissolving in
3450 ml at 20 °C and 1280 ml at 50 °C; potassium iodide may be added
to increase solubility via formation of triiodide ions (I3-).
Nonpolar solvents such as hexane and carbon tetrachloride provide a higher
solubility.
Iodine normally exists as a diatomic molecule with an I-I bond length of 270 pm,
one of the longest single bonds known. The I2 molecules tend to
interact via weak London dispersion forces, and this interaction is
responsible for the higher melting point compared to more compact halogens,
which are also diatomic. Since the atomic size of iodine is larger, its
melting point is higher.
The I-I bond is relatively weak, with a bond dissociation energy of
151 kJmol-1, and most bonds to iodine are weaker than for the
lighter halides. One consequence of this weak bonding is the relatively high
tendency of I2 molecules to dissociate into atomic iodine.
orthorhombic structure of I2
a= 0.72701, b= 0.97934, c= 0.47900 nm
|
The halogens, Cl2, Br2, and I2 adopt similar
orthorhombic structures in which diatomic molecules lie in layers:
Cl a= 0.624 b= 0.826 c= 0.448 nm
Br a= 0.667 b= 0.872 c= 0.448 nm
I a= 0.72701, b= 0.97934, c= 0.47900 nm
Return to the
course outline
or move on to Lecture 6:
Acids, Bases and Solvent Systems.
References
Much of the information in these course notes has been sourced from Wikipedia under
the Creative Commons License.
http://www.tandar.cnea.gov.ar/~gamba/z-sulfur-a/sulfurs8a.html
'Inorganic Chemistry' - C. Housecroft and A.G. Sharpe, Prentice
Hall, 4th Ed., 2012, ISBN13: 978-0273742753, pps 24-27, 43-50,
172-176, 552-558, 299-301, 207-212
'Basic Inorganic Chemistry' - F.A. Cotton, G. Wilkinson and P.L.
Gaus, John Wiley and Sons, Inc. 3rd Ed., 1994.
'Introduction to Modern Inorganic Chemistry' - K.M. Mackay, R.A.
Mackay and W. Henderson, International Textbook Company, 5th Ed.,
1996.
 Return to Chemistry,
UWI-Mona, Home Page
Return to Chemistry,
UWI-Mona, Home Page

This work is licensed under a Creative Commons
Attribution-ShareAlike 3.0 Unported License.
Created and maintained by Prof. Robert J.
Lancashire,
The Department of Chemistry, University of the West Indies,
Mona Campus, Kingston 7, Jamaica.
Created November 2014. Links checked and/or last
modified 2nd April 2015.
URL
http://wwwchem.uwimona.edu.jm/courses/CHEM1902/IC10K_MG_struct_elementsPS.html