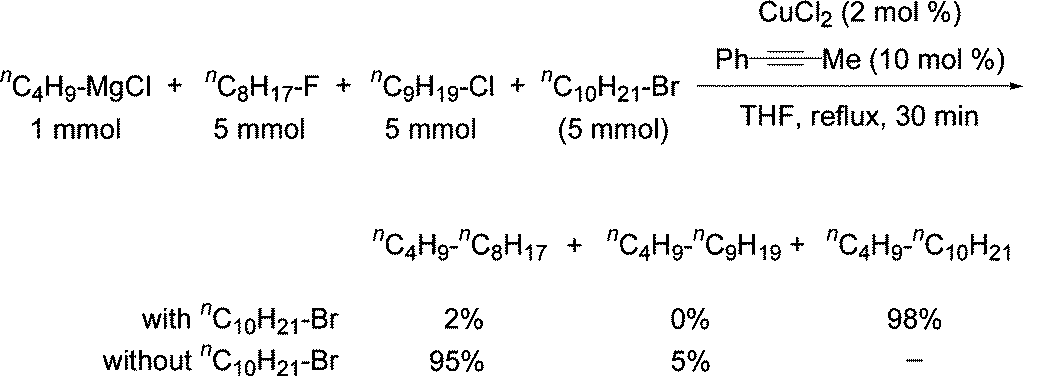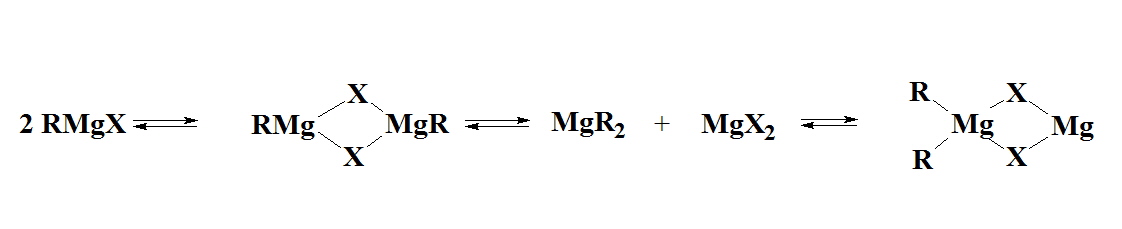
In the vapour phase Me2Be is monomeric but in the solid state it is polymeric and the bonding is considered electron deficient with 3 centre - 2 electron bonds. With higher alkyls the amount of polymerisation decreases and the tert-butyl derivative is monomeric and linear in both solid and vapour phases.
Reaction of NaCp with beryllium chloride leads to beryllocene (Cp2Be) and the solid state structure suggests that the two rings are bound to the Be differently such that 1 is designated η5 and the other η1. The experimental 1H NMR spectrum adds to the confusion of the bonding since even at 163K the protons all appear equivalent. This is accounted for by fluxional processes. Some variations of the compound have been prepared to see how general this effect is, for example, 4 protons on each ring replaced by methyl groups and all 5 protons replaced by methyl groups, (meCp)2Be, and even 4 on one ring and 5 on the other replaced with methyl groups. In the first case the fluxional process was observed down to 183K and in the second case the two rings were found to be coparallel and staggered. (Note that the structure of ferrocene is described as eclipsed when prepared at very low temperatures or in the gas phase but when formed at higher temperatures it is disordered and more staggered and since the barrier to rotation of the two rings is quite low, at 298K in the solid state there is motion).
2 K [C5Me5] + BeCl2 → (C5Me5)2Be + 2 KClA 2009 review on Grignard Reagents made
the point that over the past 100 years they had probably been the
most widely used organometallic reagents. The general procedure
for their preparation was discovered by Victor Grignard in 1900
and involved the direct reaction of magnesium with
organohalides.
R-X + Mg → R-Mg-X, (X= Cl, Br, I)
When the reaction is performed in diethyl ether or THF and in the
absence of air and moisture, the compounds are reasonably stable
although they need to be used immediately. Grignard reactions
often start slowly. As is common for reactions involving solids
and solution, initiation follows an induction period during which
reactive magnesium becomes exposed to the organic reagents. After
this induction period, the reactions can be highly
exothermic.
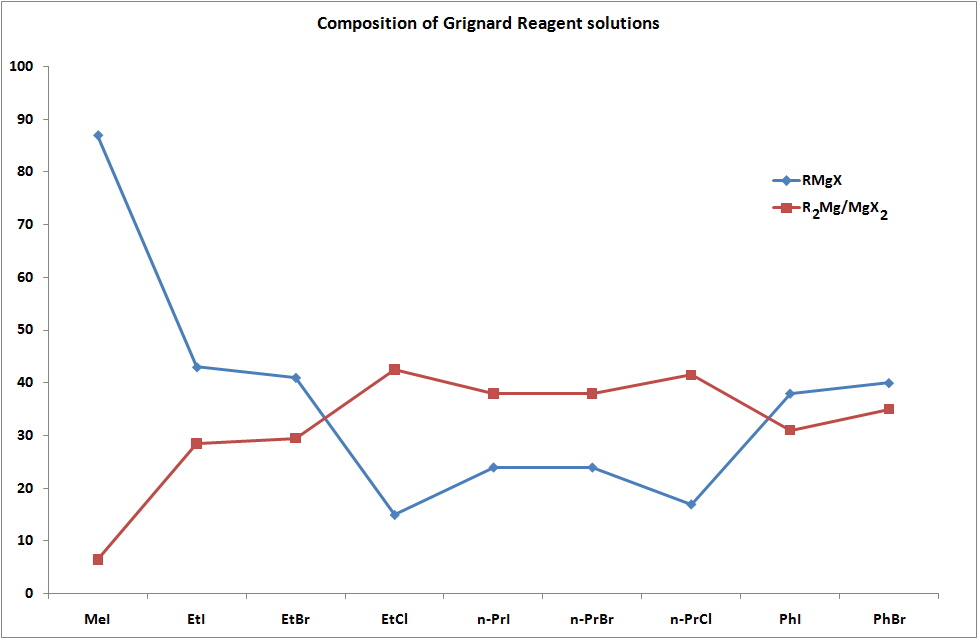
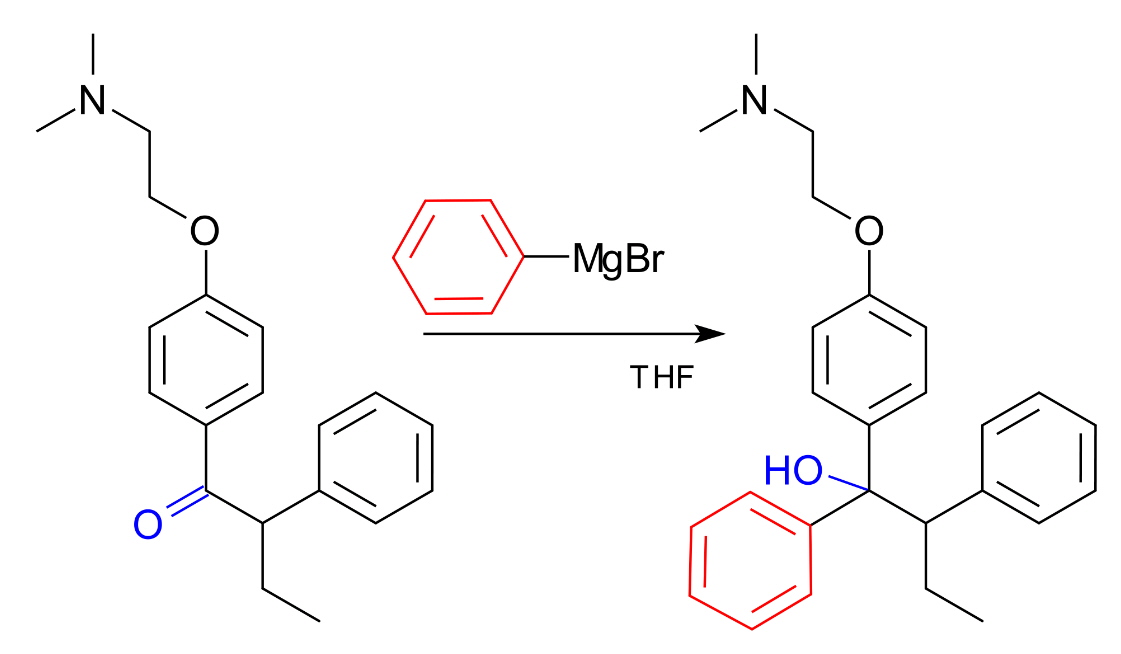
The notation of RMgX is therefore an oversimplification of what exists in ether solutions and the chart shown below indicates the percentage of RMgX for a range of Grignard Reagents.
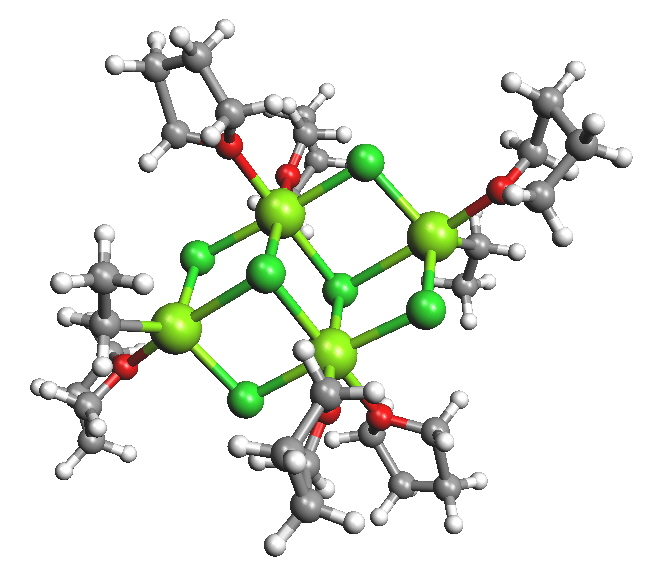
In diethyl ether, a tendency to form monomeric, dimeric and higher oligomeric species was found and was dependent on the halogen and organic substituents. In tetrahydofuran (THF) the structures were found to be closer to monomeric but it was recognised that the solid state structures may be very different to what exists in solution. For example, when "EtMgCl" is isolated from a THF solution a tetramer was found where the Mg have coordination numbers higher than the expected 4.
| metal halide | amt, mol | amt of C6H5MgI, mol | yield of biphenyl, % |
| FeCl2 | 0.01 | 0.03 | 98 |
| CoBr2 | 0.01 | 0.03 | 98 |
| NiBr2 | 0.03 | 0.095 | 100 |
| RuCl3 | 0.0036 | 0.0108 | 99 |
| RhCl3 | 0.0036 | 0.013 | 97.5 |
| PdCl2 | 0.00566 | 0.0163 | 98 |
| OsCl3 | 0.00275 | 0.007 | 53 |
| IrCl3 | 0.003 | 0.01 | 28 |
Most alkyl and aryl organoboron compounds are reasonably
stable in water, although they may still be fairly air sensitive
even pyrophoric. They are usually monomeric. For example,
triethylborane
(TEB) is strongly pyrophoric, igniting spontaneously in air.
It burns intensely with a very hot flame. The color of the flame
is apple-green, which is characteristic for boron compounds. Its
vapours may cause flash fires. This was first noted by Frankland in 1860
when he first prepared Et3B from Et2Zn by
transmetallation.
Et3B is soluble in tetrahydrofuran and hexane, and
is not pyrophoric when in solution. However the solution can
slowly react with atmospheric moisture. If the TEB solutions are
exposed to air for prolonged time, unstable organic peroxides may
form. It has been found to be toxic to the peripheral nervous
system, kidneys and testes and is extremely corrosive.
The Lockheed SR-71
strategic reconnaissance aircraft uses as fuel a mixture of
hydrocarbons known as JP-7. The very low
volatility and relative unwillingness of JP-7 to be ignited
required a pyrophoric material like triethylborane (TEB) to be
injected into the engine in order to initiate combustion and
allow afterburner operation in flight.
JP-7 jet fuel was designed to have a relatively high flash point (60 °C) to cope with the heat. In fact, the fuel was used as a coolant and hydraulic fluid in the aircraft before being burned. The fuel also contained fluorocarbons to increase its lubricity, an oxidizing agent to enable it to burn in the engines, and even a caesium compound, A-50, to help disguise the exhaust's radar signature.
JP-7 is very slippery and extremely difficult to light in any conventional way. The slipperiness was a disadvantage on the ground, because inevitably the aircraft leaked small amounts of fuel when not flying, fortunately JP-7 was not a fire hazard. When the engines of the aircraft were started, puffs of triethylborane (TEB), which ignites on contact with air, were injected into the engines to produce temperatures high enough to ignite the JP-7 initially. The TEB produced a characteristic puff of greenish flame that could often be seen as the engines were ignited. TEB was also used to ignite the afterburners. The aircraft had only 600 ml of TEB on board for each engine, enough for at least 16 injections (a counter advised the pilot of the number of TEB injections remaining), but this was considered more than enough for the requirements of any missions it was likely to carry out.
The triarylborane, BPh3, is less reactive and forms the salt Na[BPh4] that is water soluble and is useful as a precipitating agent for large metal ions.
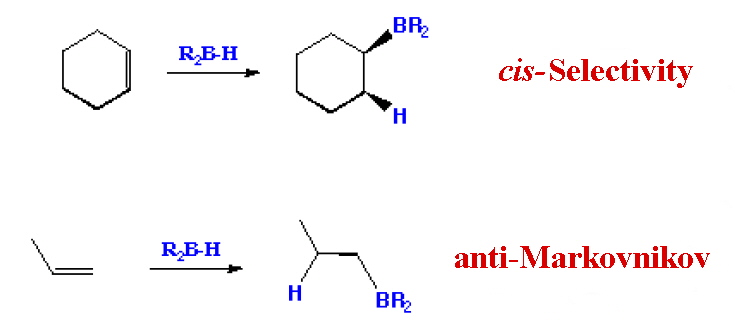
R2BCl and RBCl2 have been prepared by transmetallation and these have been used to generate species like R2B(μ-H)2BR2. One example of this is the reagent (9-BBN) that is used for the regioselective reduction of ketone, aldehydes, alkynes and nitriles. Its highly stereoselective addition on olefins allows the preparation of terminal alcohols by subsequent oxidative cleavage with H2O2 in aq. KOH. The steric demand of 9-BBN greatly suppresses the formation of the 2-substituted isomer compared to the use of borane.
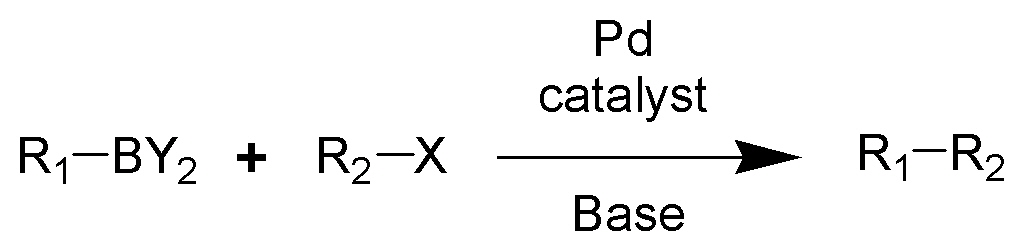
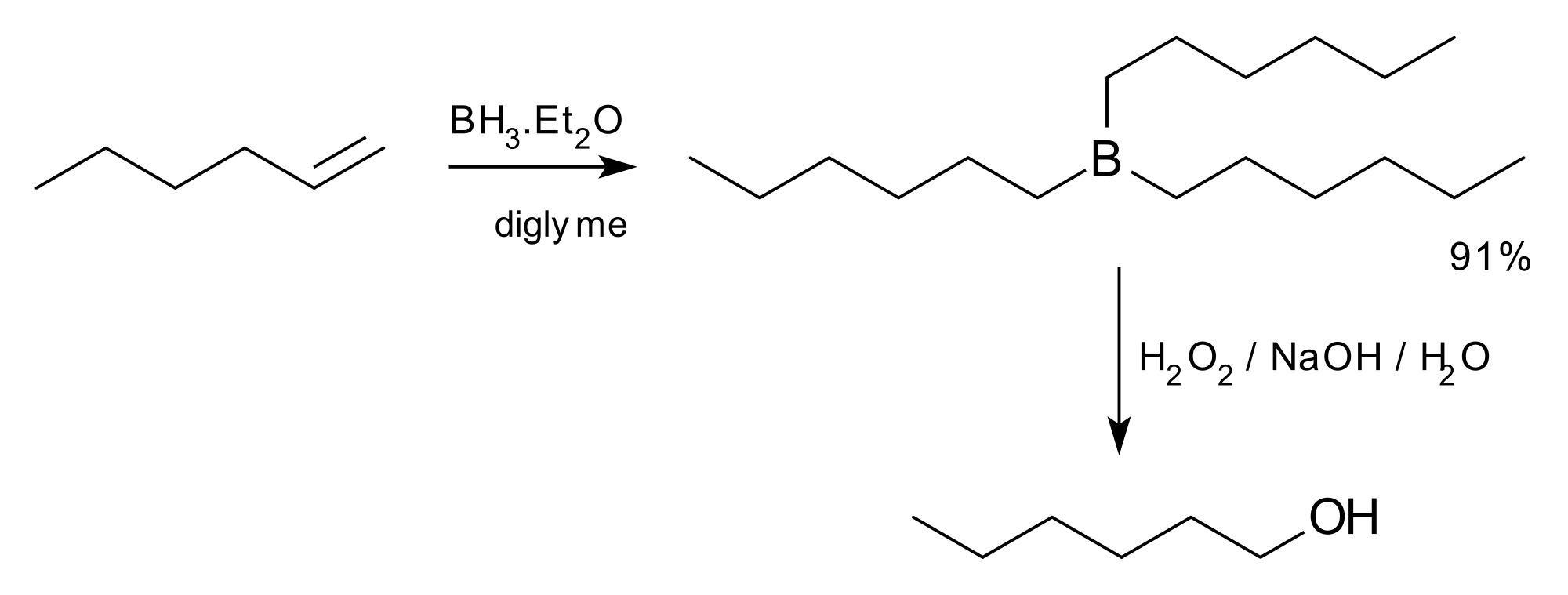
It has been found to be a useful reagent for the Suziki Reaction:The hydroboration-oxidation reaction is a two-step organic chemical reaction that converts an alkene into a neutral alcohol by the net addition of water across the double bond. The hydrogen and hydroxyl group are added in a syn addition leading to cis stereochemistry. Hydroboration-oxidation is an anti-Markovnikov reaction, with the hydroxyl group attaching to the less-substituted carbon. The reaction was first reported by Herbert C. Brown in the late 1950s and he received the Nobel Prize in Chemistry in 1979. Shown below is the original reaction described in 1957 where hex-1-ene is converted to hexanol.
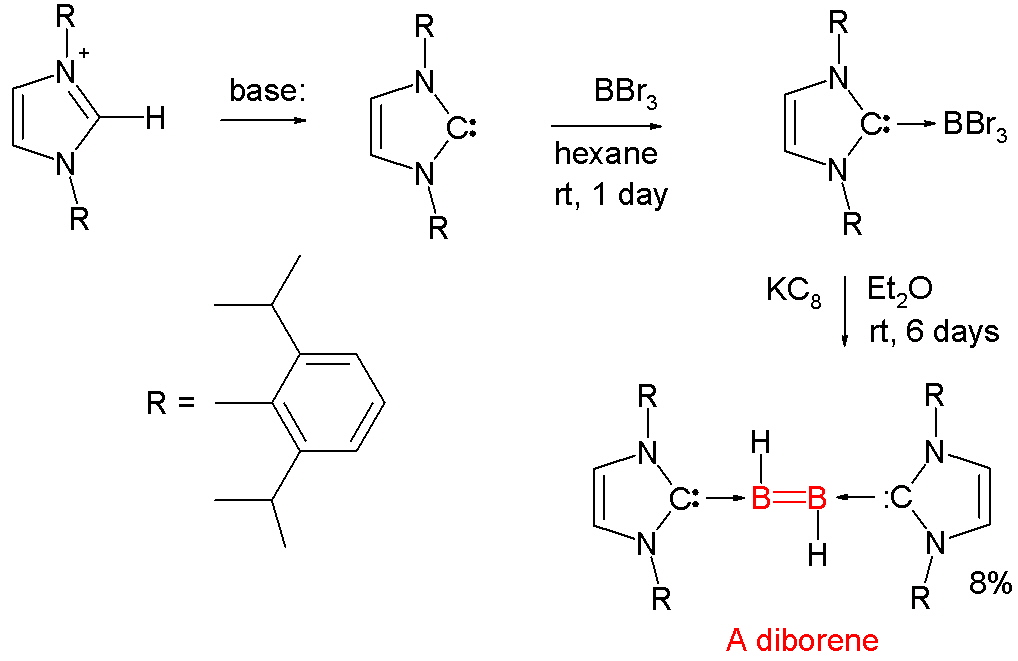
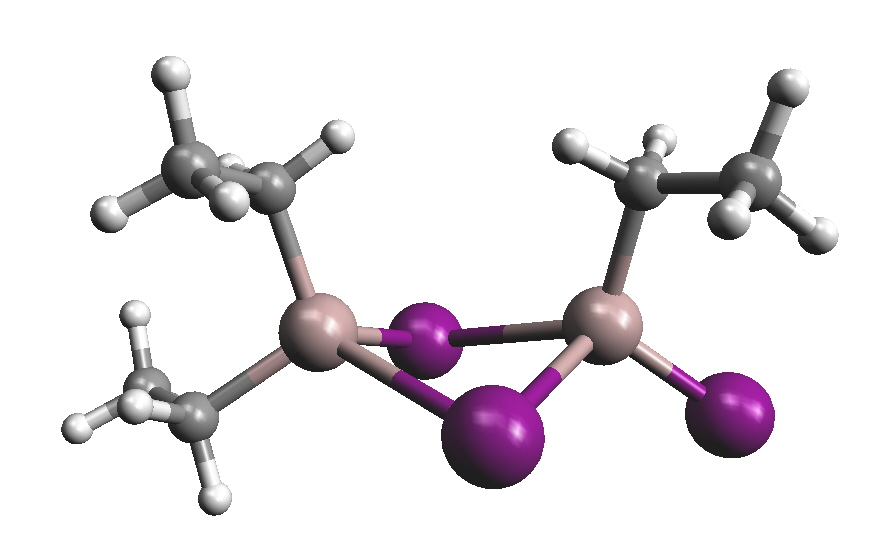
DiboreneThe first structurally characterized neutral diborene containing a B=B double bond was reported in 2007. It was prepared by the following scheme:
The first organoaluminium compound, an alkylaluminium sesqui-halide, Et3Al2I3 was reported in 1859 and was formed by reaction of elemental Al and EtI. "Me3Al" was obtained by George Buckton from aluminium and dimethylmercury as early as 1865. In hydrocarbon solution and in the solid state there is a tendency for R3Al to dimerise; this is very dependent on the size of the R group, eg a dimer for Me but monomer for tert-butyl. Me(t-Bu)5Al2 is found to be a dimer as well with the methyl group and 1 of the t-Bu groups in the bridging positions. The bridging ability is found to be Me > Et > t-Bu and clearly the case above is not what would be predicted on purely statistical terms since there are 5 times as many t-Bu groups as Me groups and there are twice as many terminal positions as bridging positions so the methyl group might have been expected to fill a terminal position.
Many organoaluminium compounds are commercially available at quite reasonable prices so that is is rarely necessary to have to prepare them in the laboratory. Reaction of Al turnings with organic halides leads to the alkylaluminium sesqui-halides. The reaction is very exothermic. The sesqui-halides do not have sharp melting or boiling points because they are in fact equilibrium mixtures:
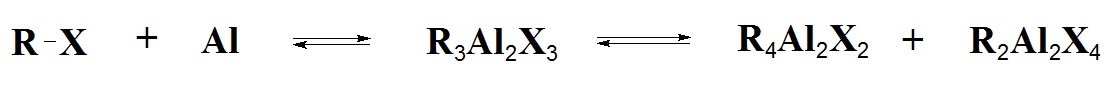
Karl Ziegler, who pioneered basic research in the field of organoaluminium compounds developed a strikingly simple yet versatile process for the synthesis of organoaluminium compounds from inexpensive starting materials. The Ziegler Direct Process allows the synthesis of triethylaluminium from aluminium metal, hydrogen and ethylene. The process involves the following:
2Al + 3H2 + 6RHC=CH2 → Al2(CH2CH2R)6 {R=H for (Et3Al)2}The Ziegler-Natta polymerisation catalysts were originally formed from Et3Al with TiCl4 and a schematic representation of the reactions at the heterogeneous surface is given below:
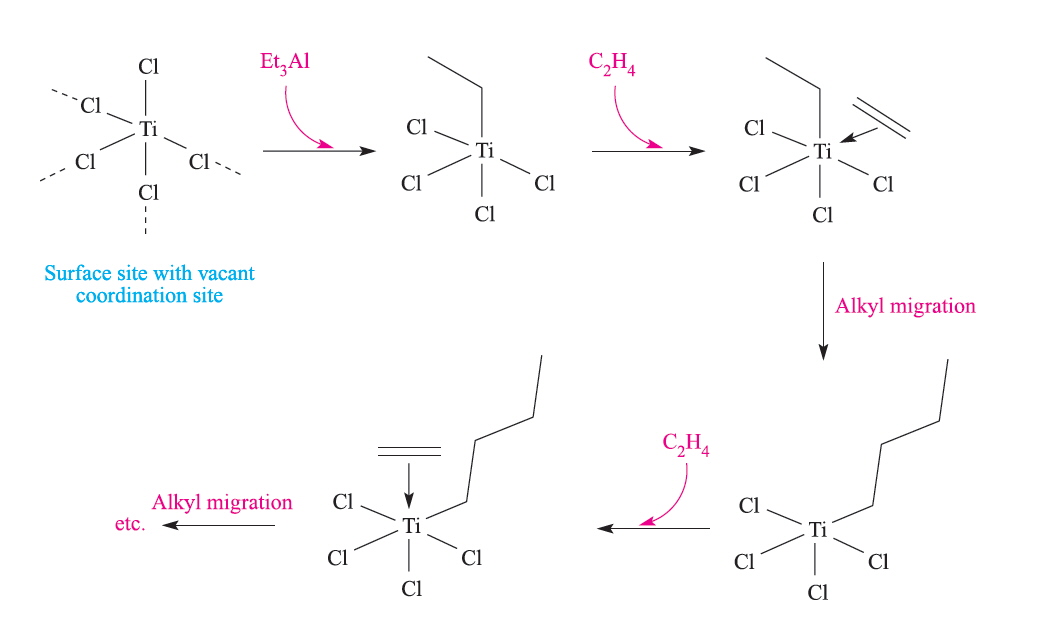
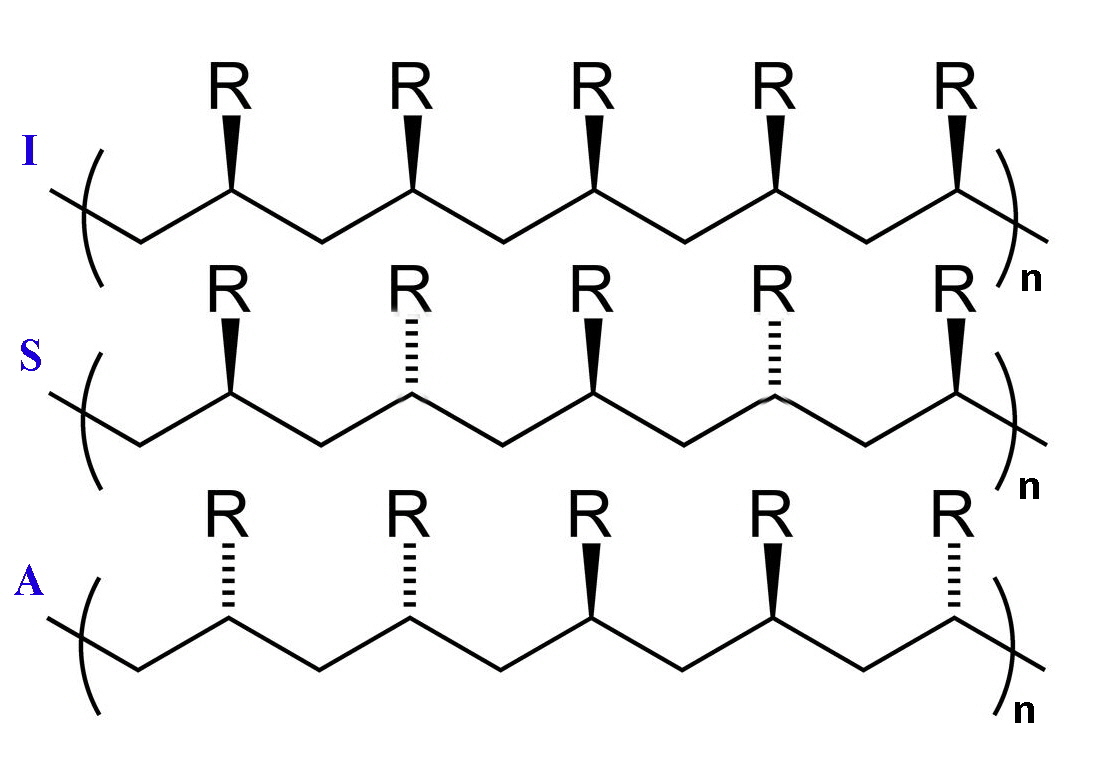
Polymerisation of ethene to high-molecular mass polyethylene occurred at relatively low pressures and the polymers were stereoregular. What this means is that isotactic polymers are formed where the R groups are all located on the same side of the carbon backbone. The resulting product gives a crystalline material since packing is more regular. The other varieties of linear polymer are called syndiotactic and atactic and in the first the R groups are on alternate sides of the carbon backbone and in the latter they are randomly distributed.
The importance of his research was soon recognized and he received the 1963 Nobel Prize for Chemistry, together with Giulio Natta who made fundamental contributions to the polymerization field.
Compounds of the type Me2Al(μ-Ph)2AlMe2 and Ph2Al(μ-Ph)2AlPh2 show that the bridging phenyl groups are almost vertical compared to the R2Al-AlR2 plane and the ipso-carbon is a distorted tetrahedral.
A similar case is found with bridging alkynes -C≡C-R where the terminal groups are t-Bu and when the Al is changed to Ga. Although in the latter case the phenyl groups are at right angles to each other.
However a very different arrangement with bridging alkynes has been found where they point towards one of the Al centres. Here the bonding is interpreted as a mixture of σ and π bonds; an Al-C σ bond connects to the first Al but the second Al centre is bound using the C≡C π bond. Examples of this are for Me2Al(PhC≡C)2AlMe2 and for the analogous Gallium compound.
 Return to Chemistry, UWI-Mona,
Home Page
Created and maintained by Prof. Robert J.
Lancashire,
Return to Chemistry, UWI-Mona,
Home Page
Created and maintained by Prof. Robert J.
Lancashire,