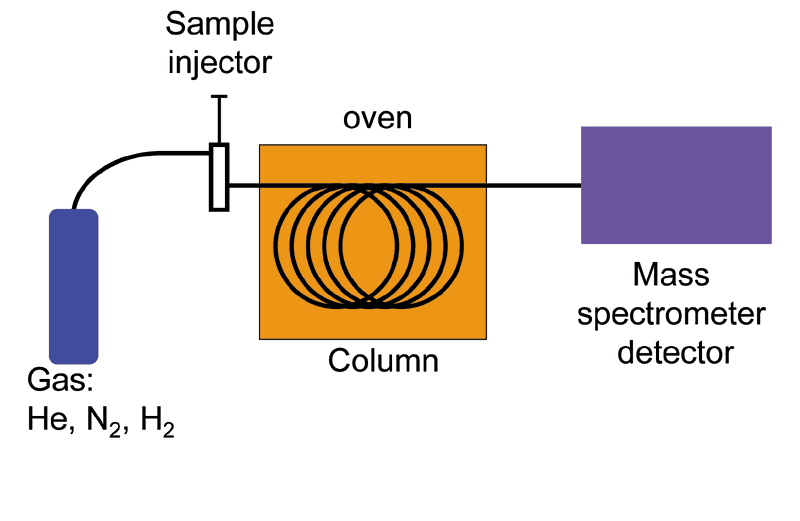
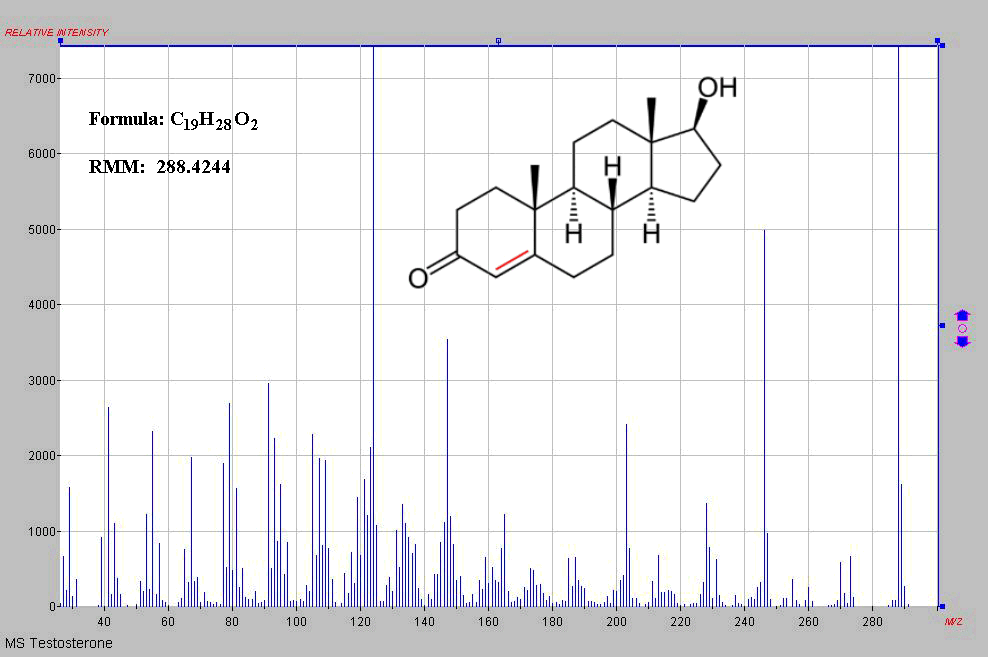
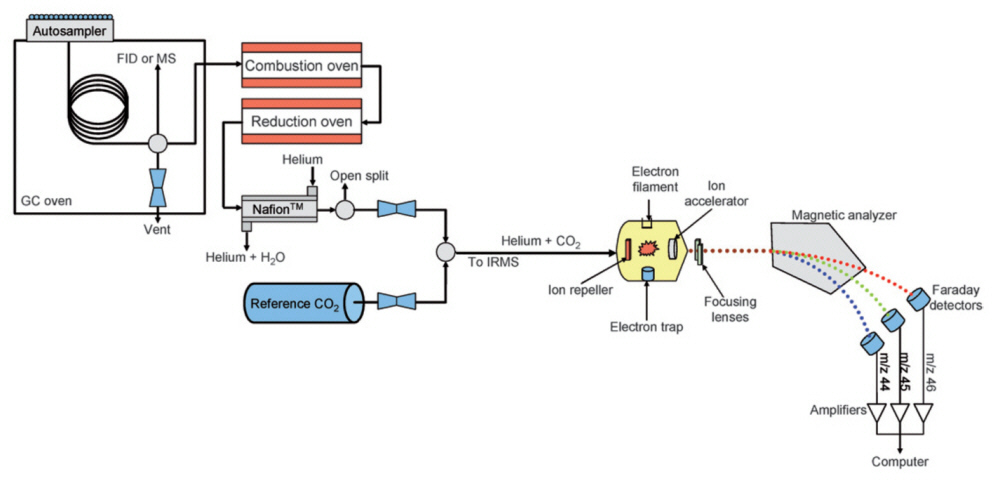
Schematic of a GCMS and the MS of testosterone

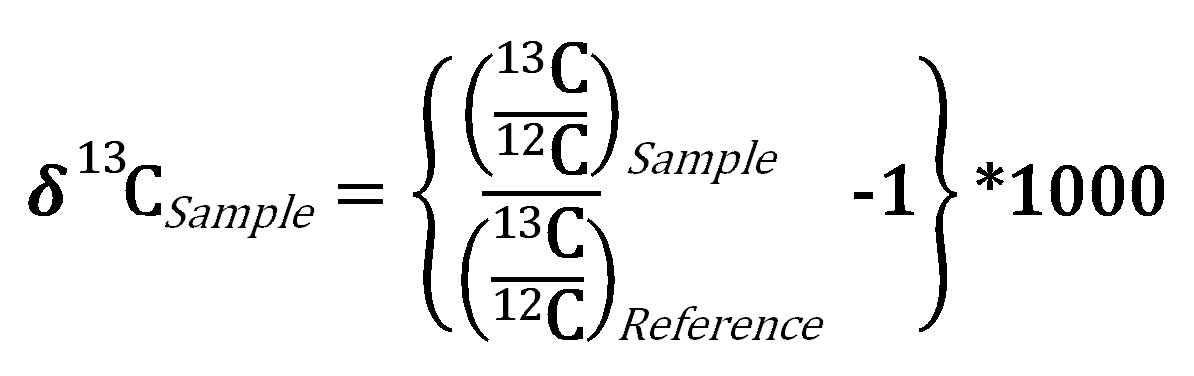
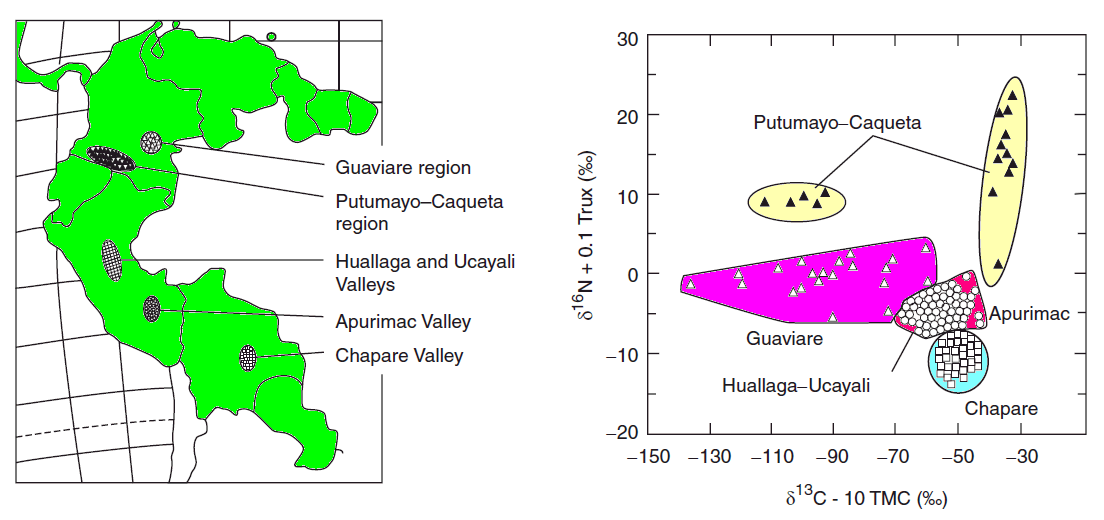
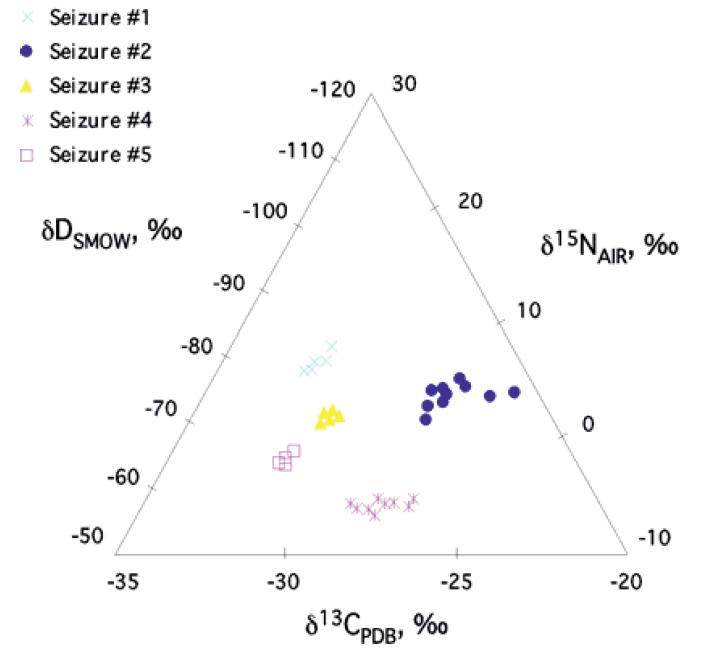
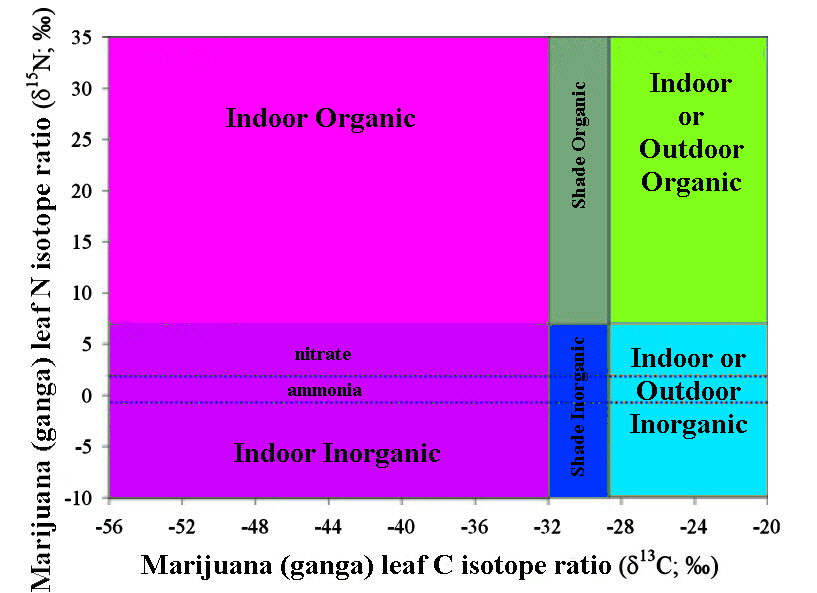
the δ13C and δ15N of 508 domestic samples of marijuana were analyzed from known U.S.A. counties, 31 seized from a single location, 5 samples grown in Mexico and Colombia, and 10 northwest border seizures. For a given subset, inflorescences and leaves were separately analyzed. These data revealed a strong correspondence, with inflorescences having slightly higher δ13C and δ15N values than leaves. A framework for interpreting the results was introduced and evaluated. Samples identified as outdoor-grown by δ13C were generally recorded as such by the Drug Enforcement Administration (DEA). DEA-classified indoor-grown samples had the most negative δ13C values, consistent with indoor cultivation, although many were also in the outdoor-grown domain. δ15N indicated a wide range of fertilizers across the dataset. Samples seized at the single location suggested multiple sources. Northwest border δ13C values suggested indoor growth, whereas for the Mexican and Colombian samples they indicated outdoor growth.

This work is licensed under a Creative Commons
Attribution-ShareAlike 3.0 Unported License.
 Return to Chemistry,
UWI-Mona, Home Page
Created and maintained by Prof. Robert J.
Lancashire,
Return to Chemistry,
UWI-Mona, Home Page
Created and maintained by Prof. Robert J.
Lancashire,
{kind=link}